Asignación taxonómica
GTDB-tk
GTDB-Tk es una herramienta que asigna taxonomía a genomas utilizando la base de datos GTDB (Genome Taxonomy Database). Basado en árboles filogenéticos y medidas de ANI (Average Nucleotide Identity), GTDB-Tk clasifica genomas bacterianos y arqueanos, proporciona una taxonomía coherente y actualizada. Se utiliza mucho en el análisis de genomas y metagenomas.
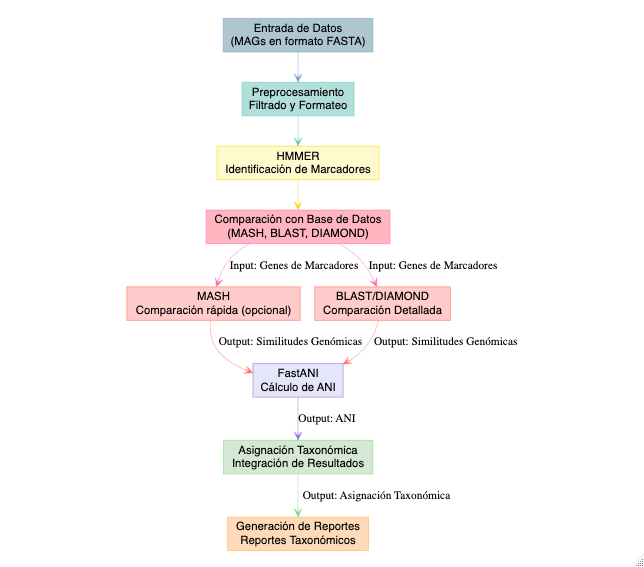
Recordemos que ya tenemos un set de bins refinados y desreplicados. Ahora vamos a asignarles identidad taxonómica, para ello vamos a correr GTDB-tk
conda activate gtdbtk-2.3.2El directorio de resultados para gtdbtk ya lo tienes en tu carpeta de resultados. Para colocar los bins refinados y renombrados ejecuta el script `src/copiar_renombrarbins.sh` :
bash src/copiar_renombrarbins.shAhora si, vamos a correr gtdbtk
nano src/10.gtdbtk.slurm#!/bin/bash
#SBATHC -J gtdbtk
#SBATCH -t 0
#SBATCH -n 4
#SBATCH -e outs/10.gtdbtk2.err
#SBATCH -o outs/10.gtdbtk2.out
#SBATCH --export=ALL
#SBATCH -p q2
#SBATCH -w compute4
# Llamamos a la base de datos
export GTDBTK_DATA_PATH=/tmp/databases/gtdb-tk/release207_v2
# ahora si corremos gtdbtk
gtdbtk classify_wf --genome_dir results/10.gtdbtk/bins/ --out_dir results/10.gtdbtk/ --cpus 4 -x fasta --mash_db /tmp/databases/gtdb-tk/release207_v2/mash/ # --skip_ani_screenSi gtdbtk está tomando mucho tiempo puedes parar el proceso con ctrl + C en tu teclado. El resultado final se encuentra en el directorio y archivo: results/10.gtdbtk/gtdbtk.bac120.summary.tsv que se copió desde el inicio.
Después de ejecutar GTDB-tk, continuaremos en R para visualizar los datos.
library(tidyverse)
library(ggplot2)
# Leer la tabla ------------------------------------------------------------####
GTDBtk <- read.table("gtdbtk.bac120.summary.tsv",
sep = "\t", header = TRUE, na.strings = "",
stringsAsFactors = FALSE) %>%
as_tibble()
# Transformar datos --------------------------------------------------------####
pozol_gtdbtk <- GTDBtk %>%
select(user_genome, classification) %>%
separate(classification, c(
"Domain", "Phylum", "Class", "Order", "Family", "Genus", "Species"),
sep = ";") %>%
rename(Bin_name = user_genome) %>%
unite(Bin_name_2, c("Bin_name", "Phylum"), remove = FALSE) %>%
select(Bin_name, Domain, Phylum, Class, Order, Family, Genus, Species)
# Guardamos los datos en un archivo de metadatos ---------------------------####
write.table(pozol_gtdbtk, file = "Metadatos.txt",
sep = "\t", quote = FALSE, row.names = FALSE, col.names = TRUE)
# Visualización de Datos ---------------------------------------------------####
GTDBtk_barplot <- pozol_gtdbtk %>%
count(Phylum, Genus) %>%
rename(Number_of_MAGs = n) %>%
ggplot(aes(x = Phylum, y = Number_of_MAGs, fill = Genus)) +
geom_bar(stat = "identity", position = position_dodge()) +
theme_minimal()
GTDBtk_barplotEn equipos revisen los resultados generados por GTDB-tk y propongan un plan para mejorar la identificación taxonómica, qué harían para darle más soporte a estos resultados?