Metabolismo
Ahora que ya tenemos los bins refinados, queremos saber qué capacidades metabólicas poseen. Para ello es necesario predecir sus genes y asignarles función.
PROKKA
Prokka es una herramienta útil, usa diferentes programas para predecir genes, secuencias codificantes, tRNAs, rRNAs. Hace la traducción de CDS a aminoácidos y asigna funciones usando varias bases de datos.
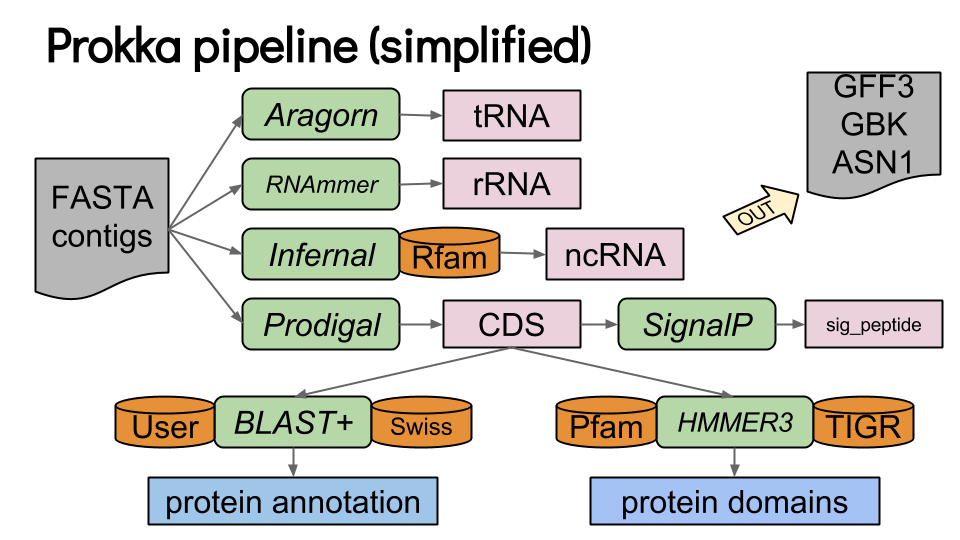
Para correrlo vamos a activar el ambiente en el que se aloja.
conda activate mt-prokkaTenemos el ambiente activo, ahora vamos a crear un directorio de resultados para prokka.
mkdir -p results/11.prokkaPara correrlo, podemos hacer un ciclo que nos permita anotar todos los bins.
for FASTA in $(ls results/10.gtdbtk/bins/); do LOCUSTAG=$(basename $FASTA .fasta); prokka --locustag "${LOCUSTAG}_Scaffold" --prefix $LOCUSTAG --addgenes --addmrn --cpus 4 --outdir "results/11.prokka/$LOCUSTAG" "results/10.gtdbtk/bins/$FASTA" ;
doneMientras prokka se ejecuta en los bins que obtuviste, despliega la ayuda y discute:
¿ qué argumentos quitarías o agregarías?
Cuáles te llamaron la atención?
Desactivemos el ambiente:
conda deactivateAhora que tenemos las proteínas predichas vamos a obtener más anotaciones útiles, usaremos kofam para esto.
KofamScan
KofamScan es una herramienta de anotación, usa la base de datos KOfam de KEGG para obtener información sobre los genes que participan en diferentes rutas metabólicas.
Creamos el script para kofam
nano src/12.kofam.slurm#!/bin/bash
#SBATHC -J kofam
#SBATCH -t 0
#SBATCH -n 4
#SBATCH -e outs/12.kofam.err
#SBATCH -o outs/12.kofam.out
#SBATCH --export=ALL
#SBATCH -p q2
#SBATCH -w compute4
# Limpiamos por si las moscas :P
module purge
# Caragmos el modulo para kofam
module load kofam-scan/1.3.0/gcc/8.3.1-n3v4
# Creamos el directorio para alojar los resultados
mkdir -p results/12.kofam
# Lo corremos
for FAA in $(ls results/11.prokka/*/*.faa); do
name=$(basename $FAA .faa)
exec_annotation $FAA \
-o results/12.kofam/"$name.txt" \
--report-unannotated \
--cpu 4 \
--tmp-dir results/12.kofam/"tmp$name" \
-p /tmp/databases/KOfam/profiles/ \
-k /tmp/databases/KOfam/ko_list
done
# remover los directorios temporales
rm -r results/12.kofam/tmp*Estos resultados ya los tienes en el directorio results/12.kofam por si esta tardando mucho.
Y ahora que ya tenemos los identificadores de KO para cada proteína, vamos a filtrar y graficar el metabolismo de los bins.
RbiMs
RbiMs es un paquete de R muy útil para obtener la anotación de cada KEGG ID y generar plots de esta información. Puede trabajar con anotaciones de KOFAM, Interpro o PFAM.
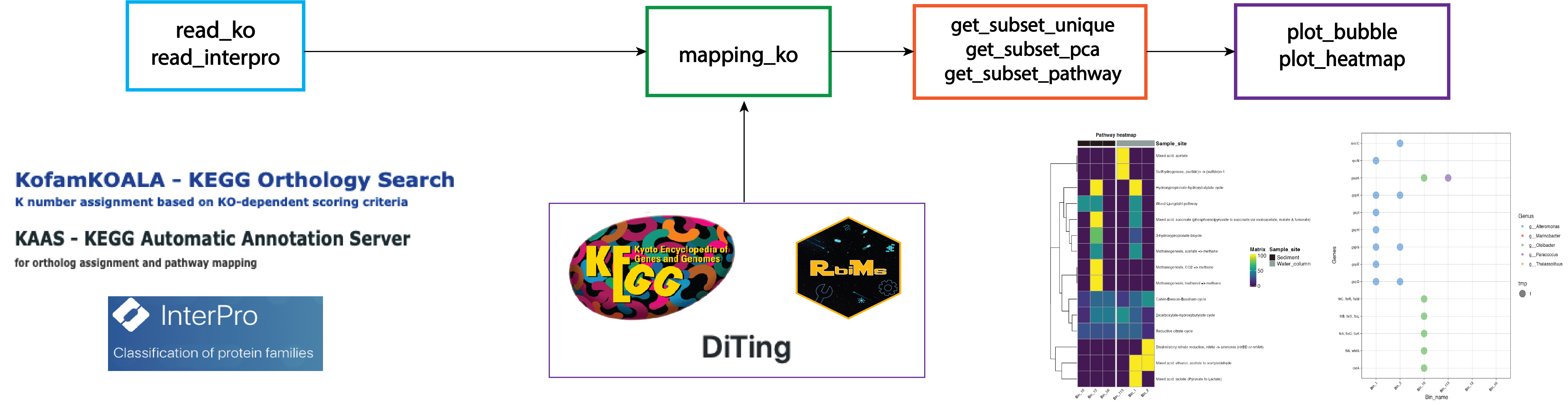
Vamos al editor de Rstudio para correr RbiMs ✨
library(tidyverse)
library(tidyr)
library(rbims)
library(readxl)
#setwd("/home/alumnoX/taller_metagenomica_pozol/")
#A continuación, leemos los resultados de KEGG
pozol_table <- read_ko(data_kofam = "results/12.kofam/")
#y los mapeamos con la base de datos de KEGG:
pozol_mapp <- mapping_ko(pozol_table)
#Nos centraremos en las vías metabólicas relacionadas con la biosintesis de aminoacidos y vitaminas:
Overview <- c("Biosynthesis of amino acids", "Vitamin B6 metabolism")
Aminoacids_metabolism_pozol <- pozol_mapp %>%
drop_na(Module_description) %>%
get_subset_pathway(Pathway_description, Overview)
#Visualizamos los datos con un gráfico de burbujas:
plot_bubble(tibble_ko = Aminoacids_metabolism_pozol,
x_axis = Bin_name,
y_axis = Pathway_description,
analysis = "KEGG",
calc = "Percentage",
range_size = c(1, 10),
y_labs = FALSE,
x_labs = FALSE)
#Añadiremos metadatos, como la taxonomía:
Metadatos <- read_delim("results/10.gtdbtk/Metadatos.txt", delim = "\t")
#Y generaremos un gráfico de burbujas con metadatos:
plot_bubble(tibble_ko = Aminoacids_metabolism_pozol,
x_axis = Bin_name,
y_axis = Pathway_description,
analysis = "KEGG",
data_experiment = Metadatos,
calc = "Percentage",
color_character = Family,
range_size = c(1, 10),
y_labs = FALSE,
x_labs = FALSE)
# Exploración de una Vía Específica
# podemos explorar una sola vía, como el “Secretion system,” y crear un mapa de calor para visualizar los genes relacionados con esta vía:
Secretion_system_pozol <- pozol_mapp %>%
drop_na(Cycle) %>%
get_subset_pathway(Cycle, "Secretion system")
#Y, finalmente, generamos un mapa de calor:
plot_heatmap(tibble_ko = Secretion_system_pozol,
y_axis = Genes,
analysis = "KEGG",
calc = "Binary")
#También podemos agregar metadatos para obtener una visión más completa:
plot_heatmap(tibble_ko = Secretion_system_pozol,
y_axis = Genes,
data_experiment = Metadatos,
order_x = Family,
analysis = "KEGG",
calc = "Binary")
plot_heatmap(tibble_ko = Secretion_system_pozol,
y_axis = Genes,
data_experiment = Metadatos,
order_y = Pathway_cycle,
order_x = Family,
analysis = "KEGG",
calc = "Binary")
# Explorar
colnames(pozol_mapp)
pozol_mapp %>%
select(Cycle, Pathway_cycle, Pathway_description) %>%
distinct()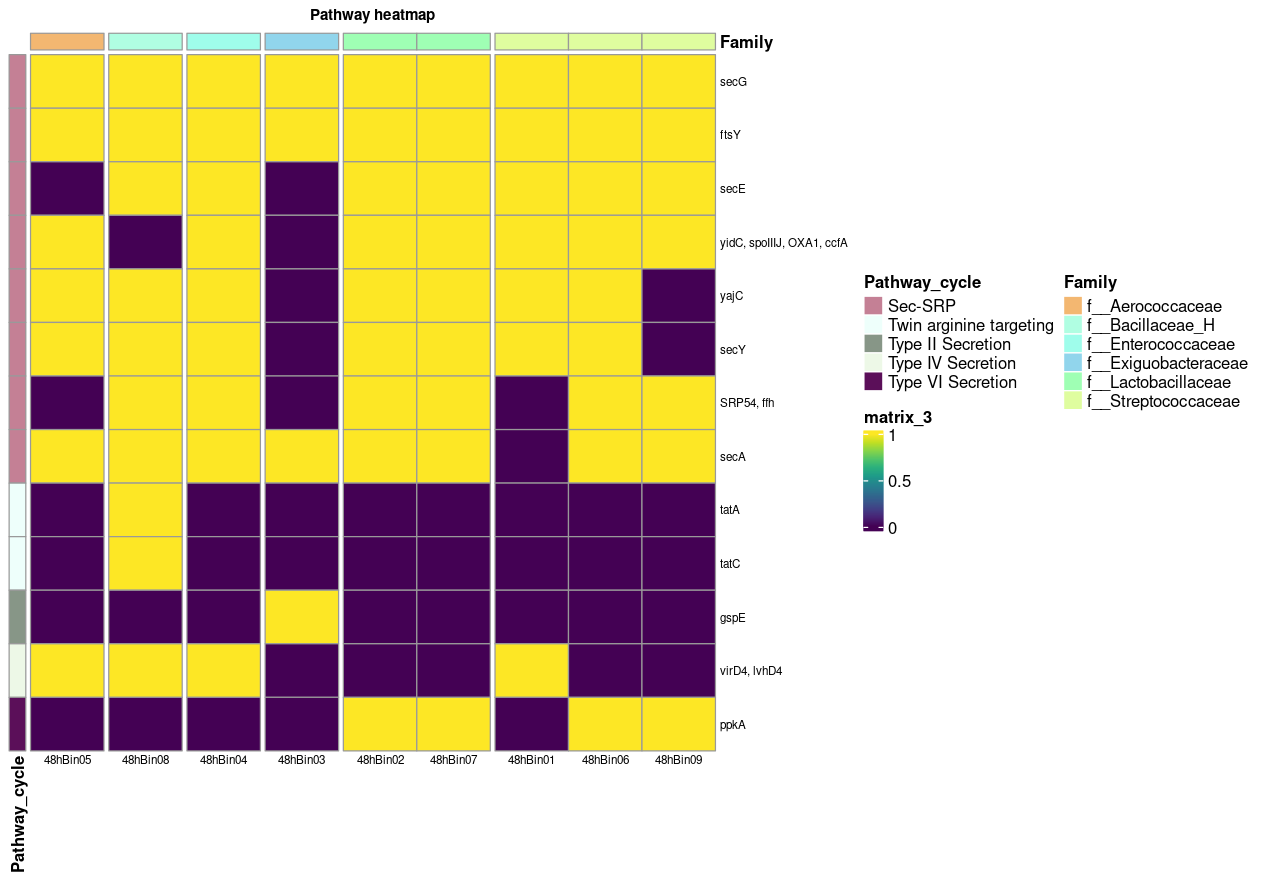
Antismash
Adicionalmente podrías anotar el metabolismo secundario de los bins siguiendo el flujo de análisis propuestos en la lección de Minería Genómica de Software Carpentry.